
This CT scan demonstrates a well circumscribed lesion in the cerebellum of this 16 year old male.
){kind=link}
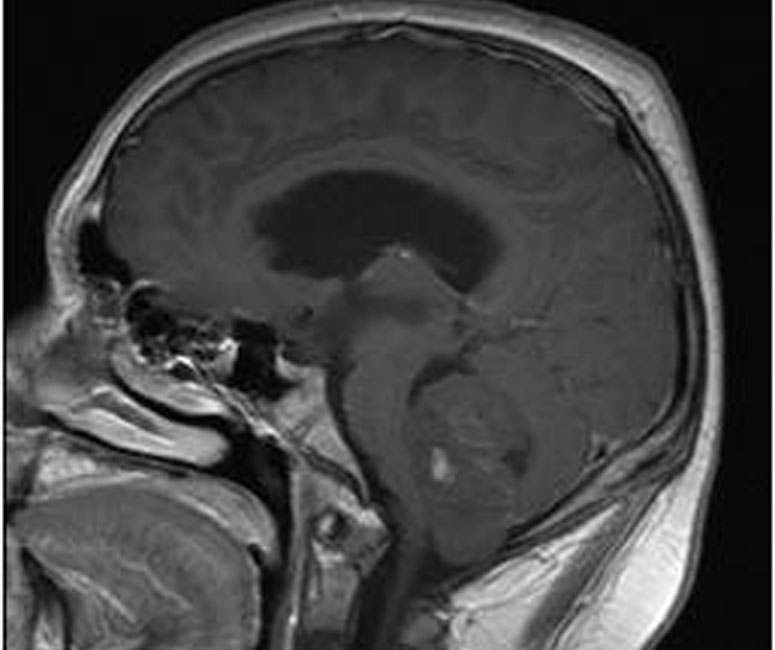
An intraventricular metastatic lesion was also identified; medulloblastoma can easily seed other parts of the CNS via the CSF.
){kind=link}
The smear from the tumor demonstrate high cellularity. The cells have hyperchromatic ovoid nuclei with scant cytoplasm.
){kind=link}
In this region, the cells exhibit fairly even chromatin and are singly dispersed. Though not well demonstrated in this two dimensional image, the nuclei have irregular membranes and have an angular to ovoid shape (Fletcher).
){kind=link}
A diffuse growth of undifferentiated cells with hyperchromatic nuclei can be seen. There are less cellular areas with increased pink fibrillary matrix.
){kind=link}
Another case demonstrating the diffuse growth of small blue round cells with admixed areas of decreased cellularity.
){kind=link}
Diffuse synaptophysin positivity supports neuronal differentiation.
){kind=link}
Strong cytoplasmic beta-catenin staining was seen in this tumor. Nuclear (not cytoplasmic) beta-catenin may predict a more favorable prognosis.
){kind=link}
Medulloblastoma is categorized under the poorly differentiated (embryonal) group of neuroepithelial tumors. Other members of this group include supratentorial primitive neuroectodermal tumor, medulloepithelioma, ependymoblastoma, cerebral neuroblastoma, retinoblastoma and atypical tertoid/rhabdoid tumor (Fletcher).
Medulloblastoma usually occurs in children and exclusively arises in the cerebellum. In children, medulloblastomas typically arise in the midline of the cerebellum whereas in the adults, the tumor tends to be more peripheral. The tumor can extend outward toward the surface of the cerebellar folia to involve the leptomeninges -- at the tumor edge, the neoplastic cells have a tendency to pallisade and invade the cerebellar cortex as a linear formation, penetrating the pia and seeding the subarachnoid space (Kumar).
Grossly, the tumor is well circumscribed with a gray color and a soft, friable texture. Necrosis is common, sometimes creating cavitary spaces; however, this is not true cyst formation as seen in pilocytic astrocytoma (Fletcher).
Histologically, a sheet of undifferentiated anaplastic cells are seen, with scant cytoplasm and hyperchromatic nuclei that may be elongated, ovoid or angulated. Mitotic activity is brisk and can be demonstrated by Ki-67 labeling; apoptotic cells are common. The tumor can express both neuronal and glial differentiation. Neuronal features include formation of Homer-Wright rosettes, fibrillarity, neurosecretory granules and immunoreactivity for neuronal markers such neurofilament epitopes and synaptophysin (Fletcher, Prayson). Glial differentiation can be highlighted by GFAP.
The desmoplastic variant comprises 10-12% of medulloblastomas and tends to be more peripheral with a much firmer consistency. Histologically, it exhibits prominent stromal collagen and reticulin deposition. There are characterisitic nodules of cells with more neuropil (and lack of reticulin) forming "pale islands" (Kumar).
The most common genetic aberration is the loss of genetic material from the short arm of chromosome 17 and occurs in about 1/3 of tumors (Fletcher).
Medulloblastoma is the most common malignant CNS tumor in the first decade of life, comprising 25% of all pediatric brain tumors. Most are diagnosed between age 5 and 10. Primitive neuroectodermal tumors (of which medulloblastoma is a subtype) comprise only 1% of adult brain tumors. Parenthetically, one must distinguish between infratentorial and supratentorial locations; the former is called medulloblastoma and the latter is called a PNET (Rieken).
The usual presentation is that of a young child with episodic vomiting and headache. Ataxia is a late presenting symptom. Rapid growth of the tumor can obstruct CSF flow, leading to increased cranial pressure, hydrocephalus, papilledema and projectile vomiting (Prayson).
Complete excision (if possible) with radiotherpay are the cornerstones of treatment.
Medulloblastomas are aggressive tumors, but are extremely sensitive to radiotherapy. It can easily disseminate through the CNS and form "drop metastases" elsewhere in the CNS. Prognosis depends on the extent of resection. Total resection plus adjuvant radiotherapy may lead to 5 year survival rates of 75%.
Unfortunately, radiotherapy in young children often leads to cognitive and physical deficits, therefore, the search continues to identify subsets of medulloblastomas with indolent behavior. In these patients, radiotherapy can be eliminated or at least tempered. For example, there is a growing body of evidence that medulloblastomas with strong nuclear beta-catenin staining have improved prognosis (Fattet, Ellison). This finding, however, does not extend to tumors with focal nuclear or cytoplasmic beta-catenin staining.
• Cerebellum : Medulloblastoma, Desmoplastic Type
Ellison DW, Onilude OE, Lindsey JC et al. beta-Catenin status predicts a favorable outcome in childhood medulloblastoma: the United Kingdom Children's Cancer Study Group Brain Tumour Committee. J Clin Oncol. 2005 Nov 1;23(31):7951-7.
Fattet S, Haberler C, Legoix P et al. Beta-catenin status in paediatric medulloblastomas: correlation of immunohistochemical expression with mutational status, genetic profiles, and clinical characteristics. J Pathol. 2009 May;218(1):86-94.
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1703-1705.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 1407-8.
Prayson R, Kleinschmidt-Demasters BK, Cohen ML. Brain Tumors. Consultant Pathology Series New York, NY: Demos Publishing: 2010: 153-5.
Rieken S, Gaiser T, Mohr A et al. Outcome and prognostic factors of desmoplastic medulloblastoma treated within a multidisciplinary treatment concept. BMC Cancer. 2010 Aug 23;10:450.
Images courtesy of Dr. Medina-Flores, Dept of Pathology, University of New Mexico.