System: Lower Respiratory : Lung: Indeterminant: Usual Interstitial Pneumonitis

System: Lower Respiratory : Lung: Indeterminant: Usual Interstitial Pneumonitis

UIP is defined as a patchy interstitial fibrosing process. Notice to the right some relatively unaffected alveolar walls compared to the marked established interstitial fibrosis on the bottom left.
This same case with older (more advanced) fibrosis in the prior slide -- the same lesion also shows young fibroplasia as seen here. There is a bluish tinge to the proliferation that appears almost myxoid.
Variable fibrosis is demonstrated again, with older (upper right) fibrosis, younger cellular proliferation (bottom) and less affected areas all mixed in together. Note the architectural distortion and the relative paucity of chronic inflammation that is typical of this condition.
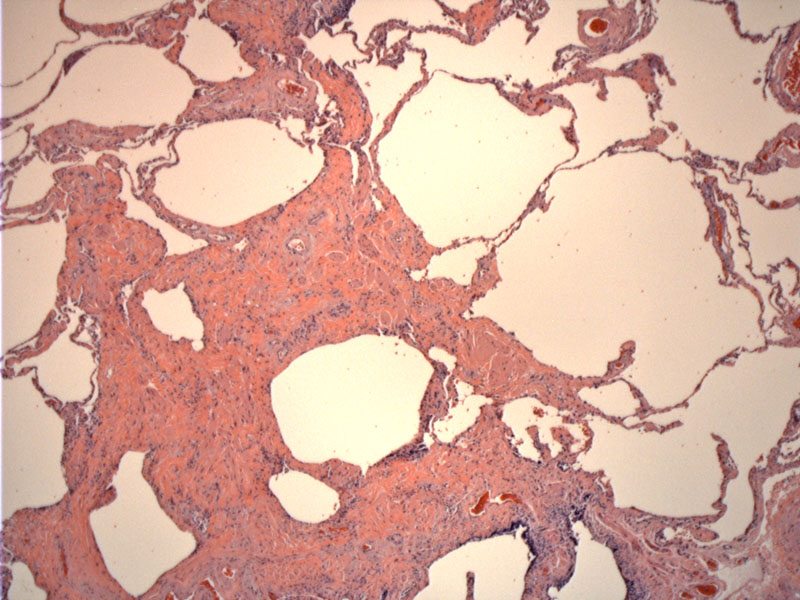
Smooth muscle proliferation is seen here, which is part of the lesion along with irregular fibrosis and cyst formation.
Diffuse parenchymal lung disease are a group of disorders involving the parenchyma between the airspaces. The categorization of these conditions has been confusing and problematic in the past and a current schema has been proposed by the American and European Thoracic Society. DPLD is divided into four major categories (Martinez):
(1) DPLD of known cause (e.g. collagen-vascular disease, drug-induced)
(2) Granulomatous DPLD (e.g. sarcoidosis)
(3) Rare DPLD with well-described clinicopathologic features (e.g. lymphangioleiomyomatosis, pulmonary Langerhan cell histiocytosis, pulmonary alveolar proteinosis, and eosinophilic pneumonia)
(4) Idiopathic interstitial pneumonia: this is further subdivided into usual interstitial pneumonia (UIP) and non-UIP. Non-UIP entities include desquamative interstitial pneumonia, respiratory bronchiolitis interstitial lung disease, acute interstitial pneumonia, cryptogenic organizing pneumonia, nonspecific interstitial pneumonia (NSIP), and lymphocytic interstitial pneumonia.
It is important to make this distinction because UIP has a poorer prognosis compared to non-UIP conditions and is usually refractory to treatment.
UIP is a chronic, progressive, fibrosing interstitial pulmonary fibrosis. Patients present with increasing shortness of breath. The disease runs an insidious course and most patients die of respiratory failure in 4-5 years. There is an association with autoimmune diseases such as SLE, RA, Sojgrens, ulcerative colitis, approximately 30% of patients have circulating ANA (Rosai). In a large portion of patients, the cause is unknown.
UIP is characterized variation in degree of infiltrate not only grossly and microscopically, but temporally as well. This leads to a patchwork appearance. In the later stages, there is irregular fibrosis accompanied by smooth muscle proliferation and formation of cysts. This lends a "honeycomb" appearance to the lung grossly.
Poor, does not respond to steroids.
Martinez FJ. Idiopathic Interstitial Pneumonias: Usual Interstitial Pneumonia versus Nonspecific Interstitial Pneumonia. The Proceedings of the American Thoracic Society 3:81-95 (2006).
Rosai, J. Rosai and Ackerman's Surgical Pathology. 9th Ed. Philadelphia, PA: Elsevier; 2004: 377-8.
){kind=link}
){kind=link}
){kind=link}
){kind=link}