System: Gastrointestinal: Liver: Benign: Polycystic Liver Disease

System: Gastrointestinal: Liver: Benign: Polycystic Liver Disease

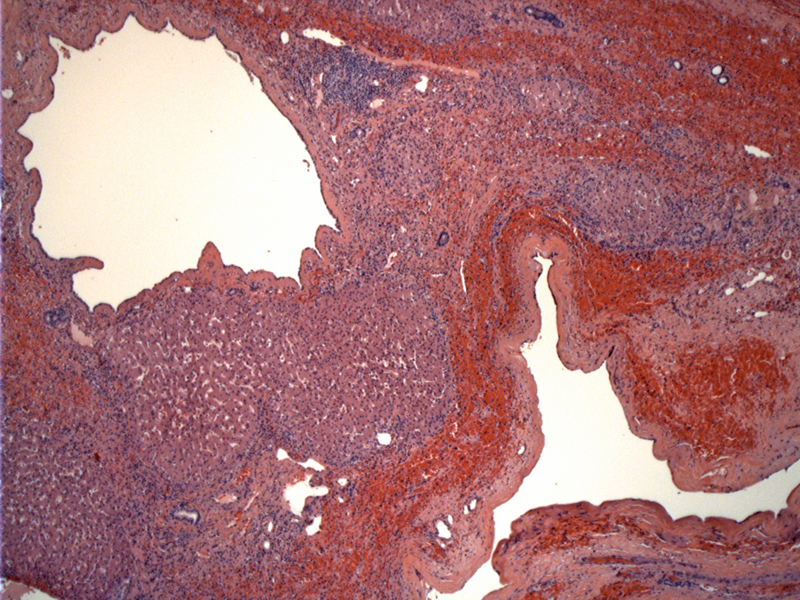
Multiple, variably sized cysts replace much of the liver parenchyma. Image
2 Image
3 Image
4 Image
5 Image
An attenuated lining is present, overlying fibrosis Image
Sometimes papillary projections are present. Pale eosinophilic fluid is found in some of the cysts. Image
Sometimes the lining is entirely denuded, replaced by inflammatory cells and hemosiderin laden macrophages. Image
9 Image
Low cuboidal epithelium consistent with ductal epithelium lines the cysts. Amorphous material is seen in the lumen. Image
multiple smaller sized cysts are seen here Image
Calcification of the wall may occur. Image
Cyst walls may collapse and eventually result in structures resembling corpora albicans of the ovary. Image
CT scan of the abdomen shows multiple too numerous to count hepatic cysts throughout the liver with small punctate calcifications intermittently. The liver is subsequently significantly enlarged, causing mass effect.
22 cm 1200 gm liver covered in cysts Image
Adult polycystic liver disease (PCLD) is an autosomal dominant condition not infrequently associated with autosomal dominant polycystic kidney disease (ADPKD). The cysts arise from intraparenchymal biliary epithelium.
Linkage to a gene on 19p13.2-13.1 has been found in association with autosomal dominant PCLD.
Although this condition is often asymptomatic, massive hepatomegaly can sometimes result in abdominal pain, early satiety, persistent nausea, dyspnea, ascites, biliary obstruction, and lower body edema.
Alkaline phosphatase, GGT, bilirubin, and AST are often normal although symptomatic patients undergoing evaluation for surgical intervention, abnormalities may be more commonly found. In this group of patients may have elevated alkaline phosphatase and GGT.
CT scan shows liver cysts with distinct margins and do not show contrast enhancement.
Complications include cyst rupture and infection.
There are associated conditions such as intracranial aneurysms and valvular heart disease,
Asymptomatic individuals do not require any intervention. Symptomatic patients are treated on an individual basis by cyst aspiration and sclerosis, open or laparoscopic fenestration, liver resection with fenestration, or liver transplantation. The surgical approach is based on the number, size and location of the cysts.
Cyst infection can have a morbidity and mortality rates of 3% and 2%. Rarely Budd-Chiari or venous outflow has been reported. Patients may develop aneurysmal diseases which may result in intracranial hemorrhage.
• Kidney : Autosomal Dominant Polycystic Kidney Disease
Cheng L, Bostwick DG, eds. Essentials of Anatomic Pathology. 2nd Ed. Totowa, NJ: Humana Press; 2006: PAGE.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: PAGE
Zhou M, Magi-Galluzzi, C. Genitourinary Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elvesier; 2006: pAGE.
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}