
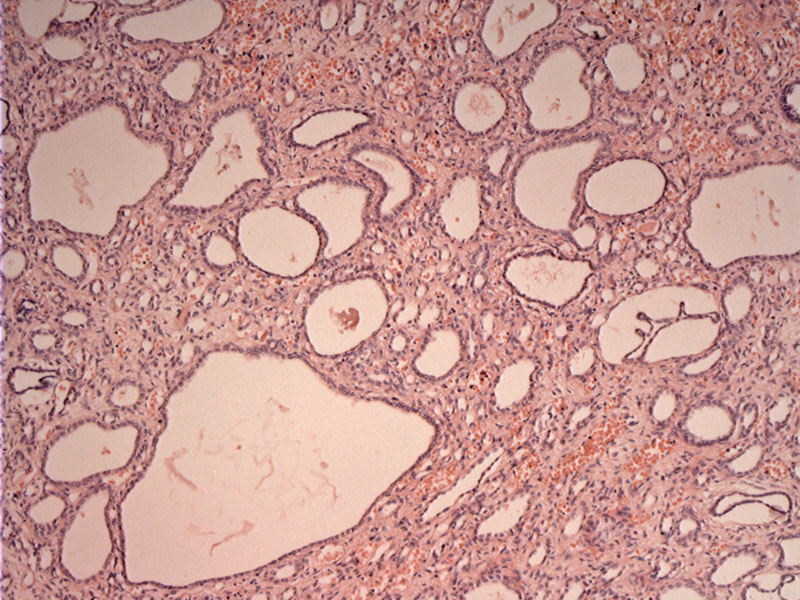
Variable-sized, large rounded cysts are seen in the medulla. This patient probably survived the neonatal period.
){kind=link}
The cysts are lined by a single layer of cuboidal cells, reflecting their collecting tubule origins. In contrast, cysts from ADPKD originate from renal tubules (Kumar).
){kind=link}
Cysts are also present in the cortex. Remember that collecting ducts are seen in both the cortex and medulla. Small functioning glomeruli are seen scattered about.
){kind=link}
Autosomal Recessive (Infantile) Polycystic Kidney Disease (ARPKD) is characterized by cystic kidneys and congenital hepatic fibrosis. In contrast to autosomal dominant PKD, this disorder is inherited in an autosomal recessive fashion.
The mutation has been localized to the PKHD1 gene encoding the protein fibrocystin located on chromosome 6p12, responsible for collecting-duct and biliary differentiation. ARPKD is categorized as perineonatal, neonatal, infantile or juvenile, depending on age at presentation and presence of hepatic fibrosis (Kumar).
Grossly, the kidneys are enlarged (200 to 600 grams) in the neonate. Numerous small cysts in the cortex and medulla impart a "spongy" appearance to the kidneys. In less severe cases (usually in older children), the cysts are larger and fewer in number -- in children, the enlargement of kidneys is not as extreme. Microscopically, there is cylindrical cystic dilation of collecting tubules. Again, in older infants and children, there are fewer cysts, but they are larger and more saccular (rounder); interstitial fibrosis and tubular atrophy and glomeruli sclerosis will be seen.
ARPKD occurs in 1:20,000 live births with a female predominance (Cheng). Clinical presentation varies, but as a very rough rule of thumb, the younger the age of presentation, the more severe the disease and renal manifestations predominate. Thus, older children have less severe renal disease, but more prominent hepatic fibrosis (Cheng, Kumar).
In the perineonates or neonates, death usually results from pulmonary hypoplasia secondary to massively enlarged kidneys or rapid renal failure. Oligohydramnios may develop from nonfunctional kidneys, leading Potter's sequence (Zhou). For those who survive beyond infancy, respiratory insufficiency does develop, but the main cause of morbidity is hepatic fibrosis, which leads to portal hypertension, esophageal varices and GI hemorrhage.
• Kidney : Autosomal Dominant Polycystic Kidney Disease
Cheng L, Bostwick DG, eds. Essentials of Anatomic Pathology. 2nd Ed. Totowa, NJ: Humana Press; 2006: PAGE.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: PAGE
Zhou M, Magi-Galluzzi, C. Genitourinary Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elvesier; 2006: pAGE.