
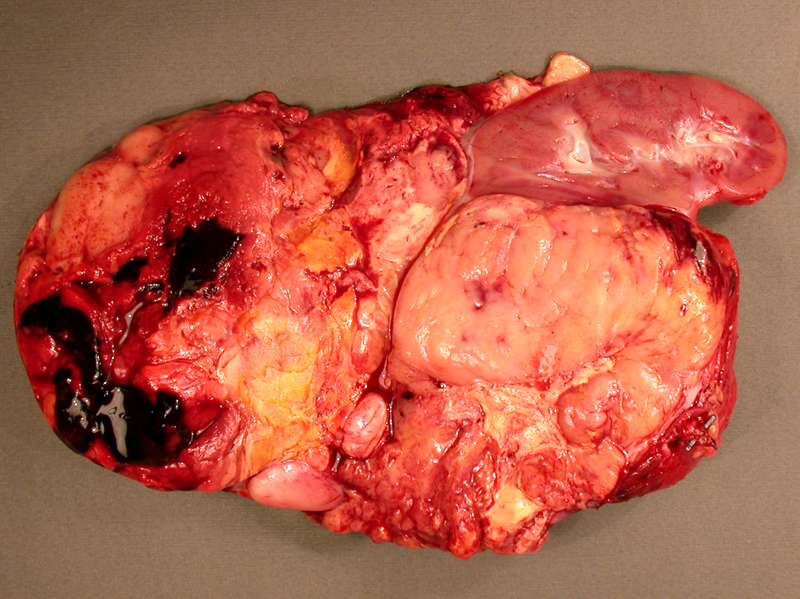
This particular ACC is a large, tan, fleshy and solid mass. Areas of necrosis, cystic degeneration and hemorrhage are common.
The tumor appear variegated, ranging from a tan-maroon to bright yellow in areas.
Microscopically, the tumor cells have a more or less uniform appearance. The cells contain a fair amount of pale to almost clear cytoplasm. From this image alone, it would be difficult to distinguish ACC from a cortical adenoma.
Necrosis is found in other regions. Confluent or widespread necrosis (when taken in combination with other findings) is supportive of a diagnosis of ACC.
Adrenal cortical neoplasms (both benign and malignant) are immunoreactive for Melan-A (seen here), as well as inhibin and calretinin. Note that not all cells uniformly stain.
A different case also shows vague nested pattern but contains marked nuclear pleomorphism.
Adrenocortical carcinoma (ACC) is rare neoplasm (1-2 cases per million) that tends to affect older individuals, but can also arise in children. Compared to cortical adenoma, ACC is more likely to be functional and thus associated with clinical manifestations of virilism, Cushing syndrome or hyperaldosteronism.
Inherited causes of ACC include Beckwith-Wiedermann syndrome and Li-Fraumeni syndrome. In Beckwith-Wiedemann Syndrome, 7.5% of children develop a malignant neoplasm, usually a neuroblastoma or ACC (Kumar, Cheng).
ACC is likely to be large (>20 cm), invasive, poorly demarcated, necrotic, hemorrhagic and cystic. Invasion into the adrenal vein and inferior vena cava is not uncommon (Kumar). Microscopically, well-differentiated ACC may be difficult to distinguish from adrenal cortical adenomas.
The modified Weiss criteria states that the presence of three or more of the following features correlates with aggressive clinical behavior (and thus, are more likely seen in adrenal cortical carcinomas): high Fuhrman nuclear grade, 5 or more mitoses/50 hpf, atypical mitotic figures, <25% clear cells, 33% or more of tumor with diffuse architecture, necrosis, venous invasion, sinusoidal invasion, or capsular invasion (Aubuert).
Peaks in the 4th decade. Typical presentation is due to hormone excess (virilism, cortisol or aldosterone excess) and possibly an abdominal mass. Metastasis to lung, liver and retroperitoneal lymph nodes may be present.
Poor -- median survival is ~ 2 years (Kumar).
→Compared to adenomas, adrenal cortical carcinomas are larger and more likely to be functional.
→Inherited causes of ACC include Beckwith-Wiedemann syndrome and Li-Fraumeni syndrome.
→ACC have strong tendency to invade into adrenal vein, inferior vena cava and lymphatics.
Aubert S, et al. Weiss system revisited: a clinicopathologic and immunohistochemical study of 49 adrenocortical tumors. Am J Surg Pathol. 2002;26:1612-1619
Cheng L, Bostwick DG, eds. Essentials of Anatomic Pathology. 2nd Ed. Totowa, NJ: Humana Press; 2006: 669-670.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: PAGE 1217-1218.
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1105-1109.

){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}