
NB classically consists of primitive-appearing, undifferentiated cells (small, blue, round cells). Note the faintly fibrillary background (neuropil) which is composed of neuritic processes of the primitive neuroblasts.
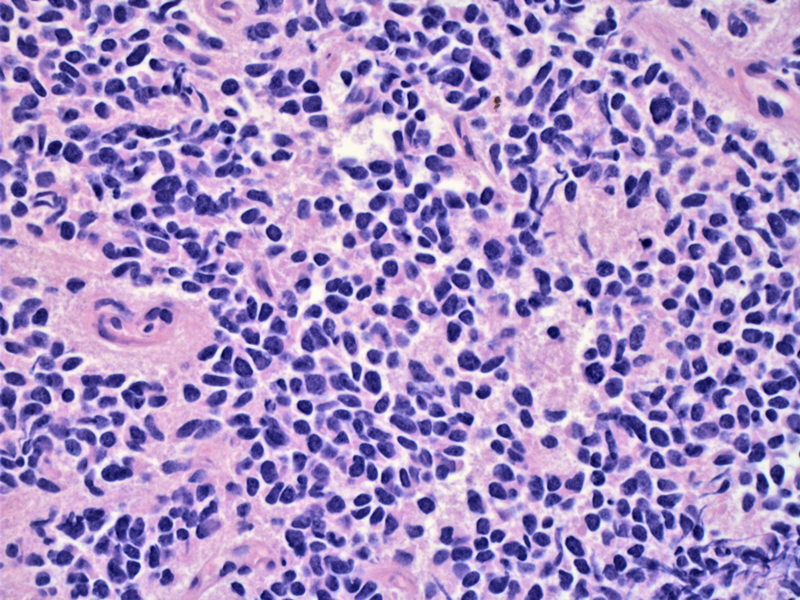
In this particular case, multiple metastatic masses were seen in the thorax. Note the diffuse growth of small round cells with dark nuclei and poorly-defined cell borders.
A Homer-Wright pseudorosette (neoplastic cells circumferentially surrounding a central area of pink neuropil) is seen just right of the center. There appears to be two population of cells, the usual neoplastic small round blue cells and a population of cells with abundant pink cytoplasm (which may either be formation of Schwannian stroma or neoplastic cells with a different morphology).
Pleural fluid demonstrates nuclei with finely stippled chromatin -- nucleoli are not prominent.
The difference between the larger mesothelial cells and admixed malignant cells can be appreciated.
On the cell block of the pleural fluid, a few small blue cells are seen, which may easily be mistaken for lymphocytes.
However, unlike lymphocytes, the cells are chromogranin positive.
They are also positive for synaptophysin.
FISH demonstrates amplification of the N-myc oncogene. A normal cell should contain only 2 copies. In this case, four pink signals are seen. N-myc amplification is currently the most important genetic test used to stratify neuroblastomas into high or low risk categories.
Another FISH image of a neuroblastoma cell with a nucleus containing 4 copies of N-myc, thus exhibiting N-myc amplification.
Other helpful immunostains may include CD56, which is strongly immunoreactive.
And as seen in the cell block, synaptophysin is diffusely positive in this histologic section.
Post chemotherapy, neuroblastomas characteristically show maturation in the form of ganglionic differentiation.
Other areas following chemotherapy exhibit dystrophic calcification, hemosiderin deposition and multinucleated giant cells.
Prognostic factors for neuroblastoma are multifactorial and complex. They include histology, age, stage as well as cytogenetic abnormalities. Note that this chart is adapted from Robbin's Pathologic Basis of Disease and Fletcher's Diagnostic Histopathology, however, it is NOT comprehensive. Expression of specific proteins such as TRK-A (tryosine kinase receptor A), CD44, telomerase and MRP (multidrug resistance associated protein) are also prognostic.
Staging chart of neuroblastomas -- note the special 4S staging.
This karyotype was read as hyperdiploid -- note the extra copies of chromosomes 6, 10, 11, 13 as well as other structural abnormalities. Hyperdiploid and near triploid chromosomal abnormalities are more favorable than diploid, near-diploid or tetraploid tumors.
The family of neuroblastic tumors are derived from neural crest cells and include three members: neuroblastomas, ganglioneuroblastomas and ganglioneuromas.
Neuroblastomas can occur anywhere along the sympathetic chain (migration pathway of primitive neural crest cells) with 70% of cases arising in adrenal medulla and intra-abdominal sympathetic chain and 20% arising in the thorax and thymus (Fletcher).
Grossly, the tumor can reach sizes of 1 kg composed of a soft, gray-tan, brain-like matter. Some masses are well-circumscribed surrounded by a fibrous pseudocapsule, but some lesions are infiltrative and invade adjacent structures such as kidneys and aorta. Larger tumors have secondary degenerative changes such as necrosis, cystic changes and hemorrhage. Calcifications are common, imparting a gritty texture and seen as punctate white-gray areas on cut section.
Microscopically, neuroblastoma is called one of the "small blue round cell tumors" for good reason. It is composed of small blue round cells set in a lightly eosinophilic fibrillary (neuropil) background - somewhat similar to cotton candy. The nuclei are finely speckled with a "salt and pepper" chromatin pattern, and nucleoli are usually inconspicuous. The cells may be separated into poorly-defined lobules or nests by delicate fibrous septae. In other instances, there may be a diffuse sheet of tumor cells without evident architecture. The neoplastic cells may form Homer-Wright pseudorosettes, when the cells arrange concentrically around a space containing neuropil (Kumar, Fletcher).
Note that the presence of (1) mature ganglion cells and (2) Schwannian stroma (organized fascicles of neuritic processes, mature Schwann cells and fibroblasts) are required before a neuroblastic tumor can be called a ganglioneuroblastoma or ganglioneuroma, entities with a more benign clinical course.
Neuroblastomas exhibit the peculiar characteristics of spontaneous (or chemotherapy-induced) maturation of primitive neuroblasts as well as spontaneous tumor regression (Kumar). IHC studies demonstrate positivity for neuron-specific enolase, S-100 (for the Schwannian components), as well as synaptophysin and chromogranin.
Neuroblastoma is the most common and more clinically important member of the neuroblastic tumors. It is the second most common malignant solid tumor of childhood (after brain tumors) and account for up to 50% of malignancies in infants. Median age of diagnosis is 22 months. There is a marginal predilection for males, and these tumors are more common in Caucasians compared to African-Americans (Fletcher)
Usually presents with a large abdominal mass, fever and weight loss. 90% of neuroblastomas produce catecholamines (similar to pheochromocytomas), which is a helpful diagnostic laboratory test. Note, however, that the catecholamines do not often cause hypertension, unlike pheochromocytomas (Kumar).
Opsoclonus-myoclonus syndrome is a paraneoplastic syndrome associated with neuroblastoma, seen in a subset of patients.
Age and stage are the most important prognostic factors. Please see our staging chart above. Infants younger than 1 year have an excellent prognosis, regardless of stage, with an estimated 5 year survival of 80-90%. Neuroblastomas in infants have a tendency to spontaneously regress. Children between 1-5 years of age have an intermediate prognosis and those older than 5 generally have a dismal prognosis.
Cytogenetics plays an important role in determining prognosis as well. Hyperdiploidy and near-triploidy corresponds with a positive response to chemotherapy and long-term disease free survival, whereas diploidy, near-diploidy and near-tetraploidy shows minimal response to therapy and generally heralds a poor prognosis.
Amplification of N-myc (an oncogene located in the short arm of chromosome 2) is present in approximately 25% of neuroblastomas, mostly in advanced disease and aggressive tumors. The more copies of N-myc, the worse the prognosis. Other important prognostic indicators include gain of a chromosome 17q and allelic loss of chromosome 1p, both of which are adverse prognostic factors (Kumar, Fletcher).
• Adrenal : Neuroblastoma (Case 2)
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1771-7.
Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005: 500-4.
***FISH and karyotype images courtesy of Skip Haines, Cytogenetics at Tricore Reference Laboratories, Albuquerque NM.

){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
.jpg', 'FISH demonstrates 4 copies of the N-myc oncogene.', 'fish', '')){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
){kind=link}
 is a favorable cytogenetic profile.', 'cytogenetics', '')){kind=link}