
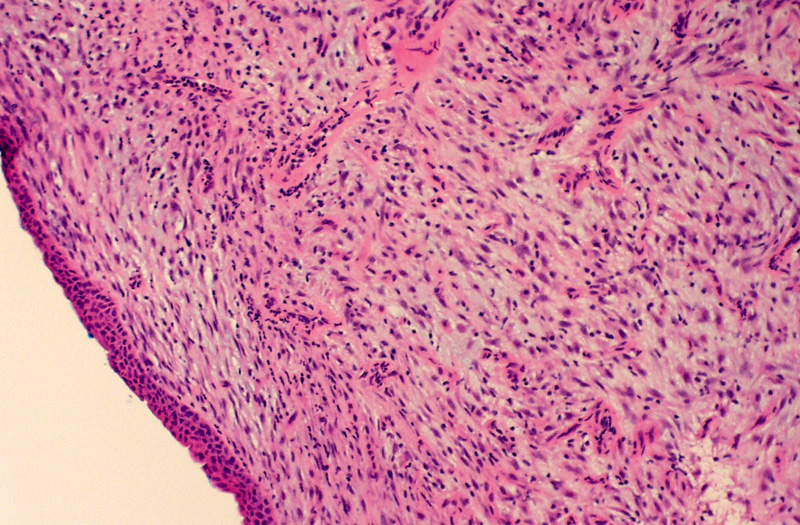
Normal bronchial mucosa is undermined by this endobronchial lesion composed of plump bland spindle cells within a myxoid stroma. A sprinkling of inflammatory cells consisting of plasma cells, lymphocytes and neutrophils infiltrates the background.
){kind=link}
This is a slightly more cellular proliferation with bland spindle cells. Again, there are scattered inflammatory cells and thin walled capillaries.
){kind=link}
Approximately 40% of cases are diffusely immunoreactive for ALK-1 as demonstrated in this tumor.
){kind=link}
This classic example of pulmonary IMT shows why the tumor was previously named "plasma cell granuloma".
){kind=link}
Inflammatory myofibroblastic tumor (IMT) is now the accepted term for lesions also formerly known as inflammatory pseudotumor, plasma cell granuloma, and inflammatory fibrosarcoma. It is a typically slow-growing tumor comprised of spindled fibroblasts and myofibroblasts in a variable background of inflammatory cells that most commonly involves the lung but has also been described in virtually any body site. Since these tumors can recur and occasionally metastasize they are considered intermediate-grade neoplasms. Although the pathogenesis was initially thought to be an exaggerated inflammatory response to tissue injury, cytogenetic and molecular studies have shown that a subset of these tumors have chromosomal abnormalities which include rearrangements involving the short arm of chromosome 2, specifically in the region of 2p23 and activation of the anaplastic lymphoma kinase gene ALK.1-3
Grossly these tumors are between 5-10 cm, well circumscribed, grey-tan or yellow, and may have a gritty texture (due to microcalcification). Necrosis or hemorrhage are rare.2
There are two histologic variants:
The plasma cell granuloma variant shows hyalinzied fibroconnective tissue with plump fibroblasts and myofibroblasts with diffuse plasma cells and admixed lymphocytes, mast cells, histiocytes, neutrophils and eosinophils.
The fibrohistiocytic variant is typically a dense spindle cell proliferation arranged in a whorled, fasicular or storiform pattern (may mimic a sarcoma).
The stroma can be hypocellular or myxoid and in older lesions there may be extensive sclerosis and hyalinization. There are usually scattered mitoses but no atypical forms. Up to 20-30% are hypercellular with focal or global atypia with necrosis, high N/C ratios, nuclear hyperchromasia and pleomorphism.1,2
The tumor cells are typically positive for vimentin, muscle-specific actin and calponin, and ALK-1 staining is seen in approximately 40% of cases. They are usually negative for desmin, caldesmon, CD34, and keratin.2
An important differential diagnostic consideration is inflammatory sarcomatoid carcinoma (ISC) which also has spindle cells arranged in a haphazard, storiform or fasicular pattern with scattered inflammatory cells. ISC typically has lymphocytes and plasma cells but usually not neutrophils, eosinophils or histiocytes. ISCs stain for EMA and keratin in contrast to IMTs which are negative for these markers.2
It is most often found in children and adolescents but is also found in all age groups with no gender predilection. The can be discovered incidentally in asymptomatic patients as a solitary, peripherally located nodule, but can also occur as an endobronchial lesion and patients may present with symptoms of airway obstruction, cough or hemoptysis. Approximately 10-20% of patients have systemic signs and symptoms such as fever, anemia, weight loss, polyclonal hyperglobulinemia and elevated ESR, which typically resolve with resection of the tumor. Radiographically these tumors are seen as a lobulated or globoid mass and sometimes with calcification.2,4
Surgical resection.
These tumors grow very slowly and there have been reported cases of spontaneous regression. A small proportion of cases may show invasion of great vessels, mediastinum, or chest wall, and local recurrence is seen in up to 25% of cases.2 The risk of metastasis is less than 5% which cannot be predicted by morphology since cytologically bland lesions can also metastasize. Extremely rare cases of malignant transformation to a high-grade fibroblastic or round cell neoplasm have been reported. Overall 5-yr survival is approximately 74% with better survival associated with completely resected tumors and tumor size less than 3 cm. 4
Coffin et al found in their study of 59 cases of IMT, ALK positivity was associated with local recurrence while ALK negativity was associated with metastasis.5
• Bladder : Pseudosarcomatous Myofibroblastic Proliferation
• Liver : Inflammatory Myofibroblastic Tumor
• Bladder : Urachal Inflammatory Myofibroblastic Tumor
• Peritoneum : Inflammatory Myofibroblastic Tumor
• Spleen : Inflammatory Pseudotumor
1 Fletcher, C. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Churchill Livingstone Elsevier; 2007: 202-203, 1553-1554.
2 Leslie KO, Wick MR. Practical Pulmonary Pathology A Diagnostic Approach. Philadelphia, PA; Churchill Livingstone; 2005: 698-702.
3 Coffin CM, Patel A, Perkins S, Elenitoba-Johnson KS, et al. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor.Mod Pathol. 2001 Jun;14(6):569-76.
4 Bootsma GP, de Bruin PC, Schramel FM, et al. Invasive inflammatory myofibroblastic tumor of the lung. J Thorac Oncol. 2009 Jul;4(7):923-6.
5 Coffin CM, Hornick JL, Fletcher CD. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases. Am J Surg Pathol. 2007 Apr;31(4):509-20.