
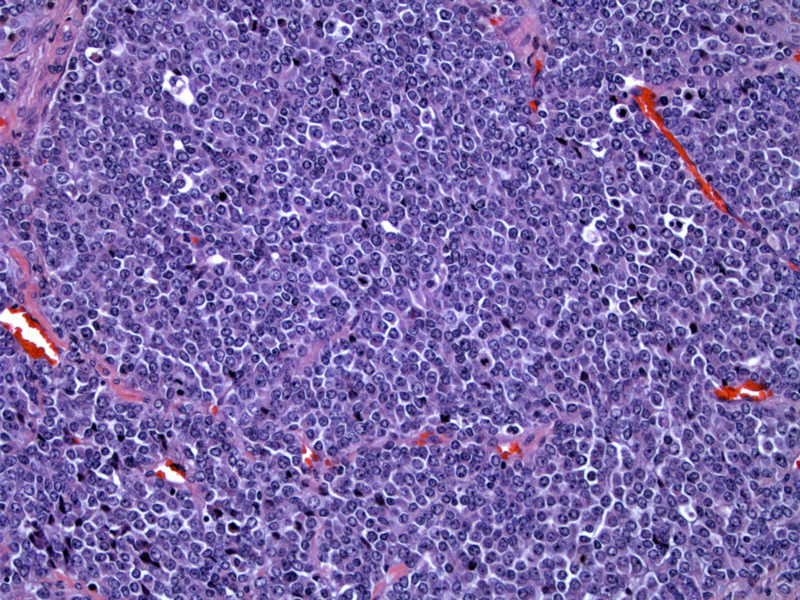
Rhabdoid tumor is composed of a sheet of monotonous polygonal cells with large vesicular nuclei and prominent nucleoli and abundant pink cytoplasm. There are numerous cells in various phases of apoptosis seen as those with darker nuclei. Mitoses are numerous.
){kind=link}
Another image demonstrates a diffuse pattern of lesional cells. This is the most common pattern, although the cells can form an alveolar or trabecular pattern.
){kind=link}
A highly characteristic feature is the presence of eosinophilic globules in the cytoplasm. These inclusions can displace the nuclei laterally, giving a plasmacytoid look.The chromatin is coarse and slightly vesicular. 3
){kind=link}
Another case is stained less heavily but also shows nice eosinophilic cytoplasmic globules. Mitotic activity is brisk.
){kind=link}
The cells are arranged in large sheets with little to no intervening stroma. Vesicular chromatin is clearly apparent.
){kind=link}
Rhabdoid tumor is the most malignant renal neoplasm of childhood, as the majority of patients (75%) die within one year of diagnosis. Fortunately, it is rare, accounting for 2% of all pediatric renal tumors.1
Grossly, the tumor is non-capsulated, poorly circumscribed with extensive hemorrhage and necrosis. The tumor tends to be located medially with involvement of the renal pelvis.
Microscopically, it is composed of monotonous polygonal cells with abundant cytoplasm. The nuclei are vesicular, spherical, with thick nuclear membranes and prominent nucleoli. The cytoplasm of the lesional cells resemble those of rhabdomyoblasts, thus the name "rhaboid" tumor. However, this tumor does NOT demonstrate differentiation toward skeletal muscle and the term is merely descriptive.
An interesting and helpful diagnostic feature is that often, one can find large globular eosinophilic cytoplasmic inclusions, which ultrastructural studies have demonstrated to be composed of tightly whorled intermediate filaments.
The cytogenetic hallmark for this tumor is the loss of hSNF5/INI1 gene on the long arm of chromosome 22.
Most patients are very young (mean age of 13 months) and these tumors are extremely rare after age 3. There is a male predominance (M:F ratio of 1.5 to 1). Most commonly presents with hematuria and hypercalcemia. The tumor is generally widely metastatic at the time of diagnosis. 15% of patients also have a primary embryonal tumor in midline posterior fossa.1,3
There is no effective treatment for this tumor.
• Uncertain Lineage : Epithelioid Sarcoma
1 Zhou M, Magi-Galluzzi, C. Genitourinary Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elvesier; 2006: 319-321.
2 Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 516-518.
3 Rosai, J. Rosai and Ackerman's Surgical Pathology. 9th Ed. Philadelphia, PA: Elsevier; 2004: 1250-1251.
Haberler C,et al. Immunohistochemical analysis of INI1 protein in malignant pediatric CNS tumors: Lack of INI1 in atypical teratoid/rhabdoid tumors and in a fraction of primitive neuroectodermal tumors without rhabdoid phenotype.Am J Surg Pathol. 2006 Nov;30(11):1462-8