
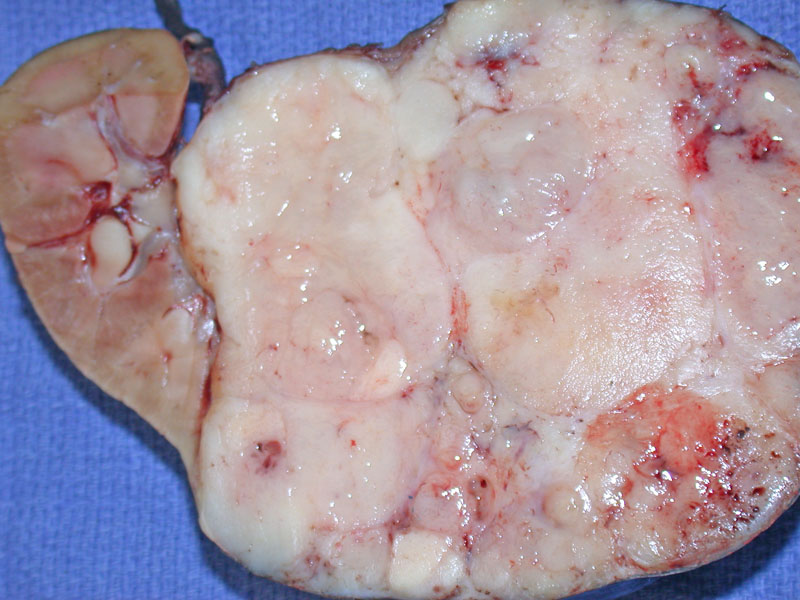
Grossly, Wilm's tumor shows a characteristic white fish-flesh appearance in the fresh state. The mass is usually large, compressing normal renal parenchyma as seen on the left. Necrosis (not shown here) can also be present.
){kind=link}
Closer examination reveals a lobular nature due to the stromal component. Again, the solid, fleshy consistency of the tumor is evident.
){kind=link}
After formalin fixation, the tumor appears more tan and less fleshy. The lobulations are more prominent as well. Note that the tumor is well-circumscribed, confined by a pseudocapsule consisting of fibrous tissue and compressed renal parenchyma.
){kind=link}
Another Wilm's tumor demonstrates prominent hemorrhage and necrosis, which obscures the mass.
){kind=link}
Wilm's tumors (nephroblastomas) classically demonstrate a triphasic histology of blastema, stroma and epithelium. However, the proportions of these components can vary widely, thus, a tumor can be biphasic or even monophasic. This image displays predominantly the blastemal component (small round or oval primitive cells with very little cytoplasm).
){kind=link}
The epithelial component is often characterized by tubule formation. However, the blastema component may blend into the epithelial cells, making it difficult to separate the two.
){kind=link}
Another image better demonstrates the tubule formation of epithelial component. There are delicate attenuated strands of fibromyxomatous stroma, which comprises the mesenchymal component of the tumor.
){kind=link}
This image demonstrates a different Wilm's tumor, which was blastemal-predominant in a diffuse pattern. The monotonous small blue cell nature of this particular tumor prompted a broader differential diagnosis, necessitating immunostains for confirmation.
){kind=link}
The WT-1 immmunostain was strongly and diffusely positive, compatible with Wilms' tumor.
){kind=link}
This image demonstrates classic Wilms’ tumor with all three cell types—blastemal, stromal, and epithelial—which can be present in various proportions. In this field, there is a preponderance ofa blastemal background with a few epithelial tubules and some stromal elements. The stromal component is appreciated slightly right of center.
){kind=link}
At times, it may be difficult to distinguish between the various elements. At this low power, the stromal and blastemal elements are difficult to separate. The stromal component may be slightly pinker.
){kind=link}
The epithelial element forming tubules is demonstrated nicely here. The background consists of blastema and stroma.
){kind=link}
Another case of blastemal-predominant Wilm's tumor demonstrates favorable histology with bland, monotonous round cells. Note the numerous mitotic figures, none of which are atypical. Features of unfavorable histology include: nucleomegaly, pleomorphism, hyperchromasia and atypical multipolar mitotic figures.
){kind=link}
A higher power demonstrates that none of the mitotic figures are atypical. A favorable histology indicates that the tumor will respond to chemotherapy. Anaplastic Wilm's tumors (unfavorable histology) are resistant to chemotherpay.
){kind=link}
Cytogenetic aberrations are nonrandom in Wilms tumor and a 2004 study of 355 karyotypes of WT found that +1q, +6, +7q, +8, +12, +13, -11 and -16 were most frequent (Hoglund). The clinical significance of these changes is still under investigation. Trisomy 10 and 12 are seen in this particular Wilms Tumor from a 2 year old boy.
){kind=link}
Anaplasia is shown in this image. Note the marked nucleomegaly with hyperchromasia in scattered cells. Anaplasia is predictive of the tumors resistance to adjuvant chemotherapy.
){kind=link}
The study of nephroblastoma (aka Wilm's tumor) reveals important information regarding the pathogenesis of tumors in general. These different genetic mechanisms for tumorigenesis are most clearly understood in the context of several hereditary tumor syndromes involving Wilm's tumors.
WAGR Syndrome: Stands for Wilm's Tumor, aniridia, genital anomalies and mental retardation. These patients carry a germline deletions of 11p13, which is where PAX6 (autosomal dominant gene for aniridia) and WT1 (tumor suppressor gene) is located. This congenital deletion of WT1 represents the first 'hit' in the two-hit theory of oncogenesis, thus, patients with WAGR have a 33% chance of developing a Wilm's tumor, as approximately a third of them eventually acquire a second 'hit'.
Denys-Drash Syndrome: These patients present with gonadal dysgenesis and early onset nepropathy characterized by diffuse mesangial sclerosis. This is due to point mutations on the WT1 (vs actual deletion in WAGR syndrome). These point mutations affect the gene product's DNA binding region.
Beckwith-Wiedemann syndrome: These patients have hemihypertrophy, organomegaly, omphalocele, and abnormally large cells in the adrenal cortex 'adrenal cytomegaly'. The genetic mutation responsible is also located on chromosome 11 at p15.5 called WT2, which is distal to the WT1 locus. The mechanism involved in oncogenesis may be due to loss of imprinting. The basis of imprinting is that only one of the parental alleles is expressed while the other allele is silenced. IGF2 is a gene product located in this region, which is a growth promoter. In some WTs, the loss of imprinting of the maternal allele causes both alleles to be expressed leading to overexpression of IGF2.1
Wilm's tumors can have a vast range of histologic appearences. Tumors exhibiting all three elements (blastemal, stromal and epithelial) are the classic "triphasic" tumors, but biphasic and monophasic tumors are not infrequent. It is important to note that each of the elements can also demonstrate different patterns and secondary differentiation.
- Blastemal: These cells are small round blue cells with darkly staining nuclei that often overlap. Frequent mitoses is common. Blastema is commonly arranged in diffuse sheets, a serpiginous pattern (with anastomosing cords) or in a nodular pattern (spherical aggregates). The serpentine and nodular patterns are helpful diagnostic features, however, if the blastema is in diffuse sheets, it may resemble numerous other entities composed of small round blue cells.
- Epithelial: Epithelial cells often have more elongated or wedge-shaped nuclei and have more cytoplasm than blastema. They are often arranged in tubules or papillary structures. Sometimes, they may even form structures resembling glomeruli.
- Stromal: The stromal element mainly consists of a fibroblastic spindle cells set in a loose myxoid background. However, these immature mesenchymal elements can differentiate into any stromal element such as smooth muscle, skeletal muscle, neuroglial tissue, fat, cartilege and bone.
Monophasic tumors may pose challenges in diagnosis. If the Wilm's tumor was composed primarily of blastema in diffuse sheets, other small blue cell tumors would enter the differential such as PNET or Ewing's sarcoma, neuroblastomas, rhabdomyosarcomas, clear cell sarcomas, synovial sarcomas and lymphomas. If the Wilm's tumor was composed of a mixture of mature stromal elements, teratomas may be on the differential.
~30% of kidneys removed for WTs demonstrate nephrogenic rests, which areas of embryonal tissue that have the potential to develop into a Wilm's tumor. Multiple foci of nephrogenic rests or diffusely distributed nephrogenic rests are termed nephroblastamosis. These nephrogenic rests may be remain quiescent, mature and become sclerotic (most common pathway), proliferate and become hyperplastic or a single cells from within the nephrogenic rest will be induced to become a Wilm's tumor. The clinical relevance of these nephrogenic rests are twofold:3
- Some of the nephrogenic rests may congregate underneath the renal cortex, forming a 'rind'. Biopsy of these rests will be virtually indistinguishable from a Wilm's tumors. Therefore, one must corroborate with radiological findings of a mass in the kidney before making a diagnosis of Wilms tumor. Other, a nephrectomy may be performed for a kidney with nephrogenic rests and no Wilms tumor.
- These rests may be found at the tumor-kidney interface, giving the appearance as infiltrating tumor.
Nephroblastoma is the most common primary renal tumor in children, with an incidence approximately 1 in 8000. The peak age of diagnosis is between ages 2-5, with 90% of tumors diagnosed before age 6. There is no gender predilection, and African-Americans have a higher incidence.4
The classic clinical scenario is that the mother or doctor notices a large abdominal mass. However, there may also be hematuria, hypertension (due to renin secretion by the tumor) and in rare instances, traumatic rupture. These tumors are generally unilateral, however, bilateral tumors arise in 5-10% of cases. A number of patients may present with pulmonary metastasis. In a child presenting with a retroperitoneal mass and pulmonary metastases, you should favor Wilms tumors over neuroblastoma.2
Pre-operative chemotherapy has been successfully applied, resulting in reduction of tumor rupture and increased favorable stage distribution. Post-operative therapy may include chemotherapy and sometimes radiotherapy in a risk-adapted approach based on histological sub-classification and tumor stage.
Tumor stage and histologic subtype are the most important prognostic factors in Wilms' tumor. More recent evidence suggests that, in addition, patient age, tumor size, response to therapy, and genetic abnormalities, specifically the loss of genetic material on chromosomes 1p and 16q, provide additional prognostic information. Survival for patients with Wilms' tumor when considered as a whole, once less than 30%, is currently greater than 90%.2 Tumors that exhibit unfavorable histology (as defined by diffuse anaplasia) have a less favorable outcome as they are more resistant to chemotherapy.
The frequency of anaplasia increases with age, as this feature is rarely seen under age 2, but is seen in 10% of tumors after age 5.3 . Anaplasia is definited as markedly enlarged nuclei (approximately three times larger than neighboring cells) and multipolar mitotic figures. WTs with anaplasia is further subclassifed into focal anaplasia and diffuse anaplasia. Focal anaplasia is anaplasia confined to discrete sites within the primary tumor and not present in extra-renal locations. In otherwords, the anaplastic regions are confined to stage I tumors and thus resectable.3
Gross images courtesy of Myra Zucker, Dept of Pathology at University of New Mexico, Albuquerque NM.
Karyotype image courtesy of Skip Haines, Cytogenetics at Tricore Reference Laboratories, Albuquerque NM.
1 Kumar V, Abbas AK, Fausto N. Robbins and Cotran Pathologic Basis of Disease. 7th Ed. Philadelphia, PA: Elsevier; 2005.
2 Rosai, J. Rosai and Ackerman's Surgical Pathology. 9th Ed. Philadelphia, PA: Elsevier; 2004:1240-5.
3 Sternberg SS, ed. Diagnostic Surgical Pathology. Philadelphia, PA: Lippincott Williams & Wilkins; 1999: 1829-1835.
4 Zhou M, Magi-Galluzzi, C. Genitourinary Pathology: Foundations in Diagnostic Pathology. Philadelphia, PA: Elvesier; 2006: 309-313.
5 Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 511-515.
6 Hoglund M, Gisselsson D, Hansen GB et al. Wilms tumors develop through two distinct karyotypic pathways. Cancer Genet Cytogenet. 2004;150(1):9-15.
Please view our case for neuroblastoma, an entity on the differential especially if the Wilm's tumor is predominately blastemal.