
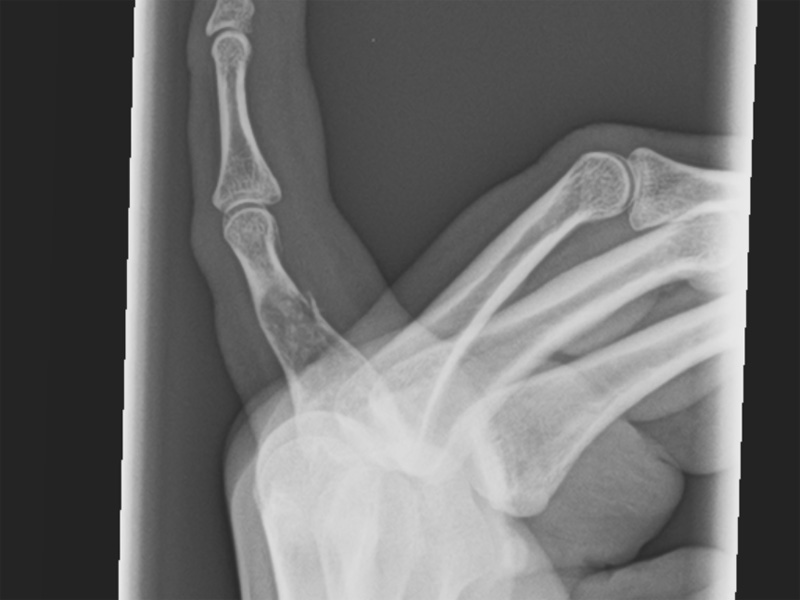
Expanded lucent area with arcs and rings on a background of more amorphous calcification.
){kind=link}
Lobular proliferation of blue cartilaginous tissue can be seen.
){kind=link}
The lesion is hypocellular with randomly distributed chondrocytes.
){kind=link}
Mild atypia may be seen. Enchondromas of the hands and feet, in particular, can exhibit increased cellularity and some nuclear atypia.
){kind=link}
Chondroma is a benign cartilaginous tumor that may arise within the bone (enchondromas), on the surface of the bone (periosteal chondroma) or in the soft tissues (soft tissue chondroma). Enchondromas most frequently arise in the tubular bones of the hands and feet. They start in the spongiosa of the diaphysis and expand outward, thinning the cortex (Rosai).
Grossly, the cut surface is rubbery with a blue clear (hyaline) surface. Histologically, there is a lobular growth pattern. Hypocellular blue hyaline dominates the picture. The transition between cartilage and the surrouding cortex is sharp. The chondrocytes are randomly scattered without the clustered or columnar formations. The rounded chondrocytes have small dark nuclei with pale pink cytoplasm.
To distinguish from a low grade chondrosarcoma, the most reliable feature is infiltration or permeation of lamellar bone by the cartilaginous tumor. Permeation of lamellar bone, however, should not be confused with ossification in an enchondroma, which is common and demonstrates a gradual transition from cartilage to bone (Folpe).
Most tend to arise in the small tubular bones of hands and feet and these can be painful due to pathologic fracture. Chondromas in the long bones are less common, and if present, tend to be heavily calcified (calcifying enchondroma). The age range is wide, but peak incidence is in the third and fourth decades with equal sex distribution (Folpe).
On imaging, characteristic "ring calcifications" represent peripheral ossification of lobules of carilage. A rim of sclerotic bone and intact cortex overlies the tumor. Soft tissue extension, transcortical spread or infiltrating growth are not compatible with enchondromas and raise the concern for a chondrosarcoma.
Most chondromas are solitary and benign, however, a subset of patients may have multiple osteomas in the setting of Ollier or Maffucci syndrome with a significant risk of malignant transformation. In Ollier syndrome (enchondromatosis), the multiple enchondromas generally arise in the first decade of life. They can occur in the small bones of the hands/feet as well as in the long tubular bones (e.g. tibia, femur). The enchondromas are asymmetrically distributed i.e. unilateral and may lead to shortening of the affected limb if the lesion develops close to and thus compromises the growth plate (Silve).
In Maffucci syndrome, multiple enchondromas are associated with soft tissue hemangiomas (e.g. cavernous hemangiomas and spindle cellhemangioendotheliomas osteomas). It is thought that 30%-50% of enchondromas in Ollier syndrome progress to chondrosarcoma (Folpe).
Both Ollier disease and Maffucci syndrome are sporadic (not familial) syndromes and it is unclear if these disorders are caused by a single gene mutation or a combination of germ-line and somatic mutations (Silve).
→Most arise in the small tubular bones of hands and feet.
→Permeation of surrounding lamellar bone is the most reliable microscopic feature in distinguishing enchondroma from a well-differentiated chondrosarcoma.
→Infiltration through the cortex into soft tissue suggests chondrosarcoma.
→Most enchondromas are benign, with the exception of those arising the setting of Ollier or Maffucci syndrome, which carry a high risk of malignant transformation.
• Chondroid : Chondromyxoid Fibroma
• Chondroid : Chondrosarcoma, Conventional Type
Fletcher CDM, ed. Diagnostic Histopathology of Tumors. 3rd Ed. Philadelphia, PA: Elsevier; 2007: 1604-5.
Folpe AL, Inwards CY. Bone and Soft Tissue Pathology: Foundations in Diagnostic Pathology Philadelphia, PA: Elsevier; 2010: 339-342.
Rosai, J. Rosai and Ackerman's Surgical Pathology. 9th Ed. Philadelphia, PA: Elsevier; 2004: 2158.
Silve C, Juppner H. Ollier Disease. Orphanet J Rare Dis. 2006;1:37.